www.joiiup.com
粒線體功能障礙與慢性疾病關係的醫學論證(下)



IV. 粒線體功能障礙在代謝性疾病中的作用
A. 粒線體:代謝的核心
你可能聽過粒線體是「細胞的發電廠」,但它其實不只會發電,還是整個身體代謝的關鍵控制中心。在每一個細胞裡,粒線體負責將食物中的主要營養成分——葡萄糖、脂肪酸、胺基酸——進行氧化,轉換成細胞活動所需的能量(ATP)[12]。
這些能量支持你呼吸、跳躍、思考、修復組織,甚至只是坐著不動也需要!
除了幫助產生能量,粒線體還參與了許多與慢性病密切相關的代謝過程,例如:
調節血糖與胰島素分泌:
在胰臟的 β 細胞中,粒線體會感應血糖變化並驅動胰島素的釋放。這是控制血糖的第一道防線。
增加胰島素敏感性:
在骨骼肌、肝臟和脂肪組織裡,粒線體的運作會影響細胞對胰島素的反應程度 [13]。換句話說,粒線體健康越好,胰島素調控越有效,越不容易發展成第二型糖尿病或代謝症候群。
從能量產生、血糖控制,到脂肪代謝與抗發炎反應,粒線體幾乎參與了你體內所有的「後勤運作」。當它們健康、運作正常時,你的身體就能有效應對日常活動與壓力;但一旦粒線體功能失調,代謝問題、胰島素阻抗甚至慢性疾病就可能悄悄上身。
B. 第二型糖尿病(T2D)與胰島素阻抗(IR):複雜的相互作用
1. 大量研究證實,粒線體功能障礙與 T2D 和 IR 密切相關 [13]。這種關聯在多個關鍵代謝組織中都有發現,包括骨骼肌、肝臟、胰腺 β 細胞和脂肪組織 [13]。某些特定的 mtDNA 突變甚至可以直接導致糖尿病,稱為粒線體糖尿病 [10]。
2. 在胰島素阻抗中的機制(尤其骨骼肌):
OXPHOS/底物氧化能力下降: 在第二型糖尿病(T2D)或胰島素阻抗(IR)的人身上,骨骼肌的代謝能力會變差,其中一個重要原因就是粒線體功能下降。具體會發生什麼?
▼ 粒線體變少:就像工廠縮編,能量製造單位不夠用
▼ 氧化酶活性變弱:等於設備老舊,發電效率降低
▼ 粒線體DNA變少:影響粒線體的正常維護與複製
▼ 粒線體的建造與調控失常:關鍵調節蛋白 PGC-1α 表現下滑,導致整個「建廠系統」卡住 [29]。
這些改變讓骨骼肌沒辦法有效利用脂肪酸或葡萄糖來產生能量 [13]。
更麻煩的是,肌肉原本應該根據身體狀況靈活切換能量來源(例如從「燒糖」轉成「燒脂肪」),但現在這個切換系統壞了,科學家稱之為:代謝不靈活性(metabolic inflexibility)[30]。也就是說,當你吃飽、運動或進入休息狀態時,肌肉無法快速調整用哪一種燃料,導致能量使用效率低落,進一步加重胰島素的工作負擔,形成惡性循環。
胰島素阻抗不是單靠胰島素本身的問題,也跟粒線體的健康有很大關係。尤其在骨骼肌中,當粒線體「數量不足、效率不佳、調控失靈」,整個身體的代謝彈性就會被拖垮,最終讓血糖與脂肪都「不受控」。
● 脂毒性(Lipotoxicity)
這種現象當身體無法有效燃燒脂肪,特別是在骨骼肌或肝臟等重要組織中,脂肪酸會在細胞內不斷堆積,變成一種隱形的威脅。
當粒線體的脂肪酸氧化能力下降(也就是脂肪「燒不完」時),多餘的脂肪酸不會馬上排掉,而是轉化成一些具有生物活性的脂質中間產物 [13],例如:甘油二酯(DAGs)與神經醯胺(Ceramides)。
這些不是「普通的脂肪」,而是會主動干擾細胞功能、破壞胰島素作用的有害分子。這些脂毒性分子會:
▼ 抑制胰島素訊號傳導路徑 ——例如阻礙 IRS(胰島素受體底物)與 Akt 的磷酸化作用,使訊號無法順利傳遞 [29]
▼ 影響 GLUT4 轉運蛋白 ——特別是當神經醯胺累積在粒線體內,會阻礙 GLUT4 移動到細胞膜,讓葡萄糖無法順利進入細胞 [31]。
結果就是:
血糖降不下來,胰島素用力過猛也沒用,形成典型的「胰島素阻抗」。當粒線體無法有效「燒脂肪」,脂肪不但囤積,還釋出會破壞訊號系統的「毒性脂質」,讓原本應該幫助控糖的胰島素也跟著失靈。
所以不是你吃太多油,而是細胞不會處理油了!
守住粒線體的脂肪代謝能力,也是在守住你的血糖穩定力!
● 氧化壓力
當粒線體出現功能障礙時,它們不僅「發電發得不好」,還會產生過量的自由基(稱為活性氧,ROS)。這些過量的自由基就像細胞內的「化學煙霧」,會干擾正常運作,甚至破壞細胞訊號系統。當 ROS 累積過多時:
▼ 攻擊胰島素訊號分子:自由基會氧化細胞內的關鍵蛋白,使它們無法正確地接收與傳遞胰島素的訊息。
▼ 啟動壓力激酶(如JNK):這些「壓力酶」就像警報器被誤觸,會進一步抑制胰島素路徑中的關鍵節點,讓胰島素訊號更難傳下去,進一步加劇胰島素阻抗(IR)[13]。
除了自由基之外,功能異常的粒線體還會導致一種能量代謝失衡:當細胞內 NADH/NAD⁺ 比例過高,代表電子堆積、能量流通不順,這不但會增加 ROS 生成,還會干擾脂肪的 β -氧化(燃燒脂肪的過程),進一步促進脂肪堆積與胰島素阻抗 [32]。
粒線體若無法妥善運作,就會:發電減少、自由基激增、激活壓力酶及干擾脂肪代謝,最終讓胰島素「喊破喉嚨都沒人聽」。所以,降低氧化壓力、維持粒線體平衡,是改善胰島素阻抗的重要策略之一。
● 動力學改變
在糖尿病患者的肌肉細胞中,研究發現粒線體的形狀和排列方式出現了變化:它們不再是健康、有彈性的網絡,而是變得破碎、零散,就像一堆「迷你發電碎片」。
這種現象被稱為粒線體碎片化,是粒線體動力學失衡的表現——也就是:分裂太多,融合太少。碎片化的粒線體會出現兩大問題:
▼ 產生更多自由基(ROS):這會傷害細胞內的胰島素訊號傳導機制,導致「胰島素叫不動細胞」。
▼ 影響粒線體品質與修復:原本融合是粒線體自我修補的機制,現在無法修補,只能繼續壞下去。
結果就是:胰島素效果變差、血糖難控制,形成惡性循環。研究顯示,高血糖本身就會促進粒線體的分裂 [13]。也就是說,血糖越高,粒線體越碎;粒線體越碎,胰島素越無效,血糖就更高,這是一個典型的代謝惡性循環。
粒線體不是靜態的發電機,而是需要維持動態平衡的「細胞網絡」。而在糖尿病中,這個網絡被高血糖打亂,變得碎片化,進而破壞細胞代謝與胰島素反應。所以,恢復粒線體的融合分裂平衡,不只是「修發電廠」,更可能是解決胰島素阻抗的關鍵!
3. 在 β 細胞功能障礙中的機制 [5]:
胰島素分泌受損: 胰島素的分泌過程其實是一場高度依賴能量的反應,而這份能量正是由粒線體負責提供的。胰島素是怎麼被分泌出來的?當我們吃進糖分(葡萄糖),血糖上升,胰臟裡的 β 細胞就會啟動一連串「產能→訊號→釋放」的程序:
1. 葡萄糖進入細胞
2. 粒線體開始燃燒葡萄糖,產生ATP(能量分子)
3. ATP會關閉鉀離子通道(KATP通道)
4. 這會讓細胞膜產生電位變化(去極化)
5. 鈣離子進入細胞,引發胰島素儲存囊泡釋放
6. 胰島素進入血液,幫助細胞吸收葡萄糖,血糖下降
當粒線體功能出現障礙,例如:mtDNA突變(粒線體內的基因受損)及長期處於高糖高脂環境(飲食不當)。這會讓粒線體產能不足,ATP 量不夠,KATP 通道關不起來,電訊號發不出來,胰島素也就出不來了。
結果就是:吃進糖分後,身體「沒能量啟動胰島素反應」,血糖就一路飆升。
粒線體在胰島素分泌中扮演著「供電總機」的角色。如果粒線體電力不足,胰島素就無法即時釋放,等於血糖控管失靈,為第二型糖尿病鋪路。保護粒線體,就是守住胰島素反應力!
▼ ROS增加/細胞凋亡:β細胞「過勞死」的真相
胰島素是由胰臟中的 β 細胞分泌的,而這些 β 細胞,就像是身體裡的血糖守門員,負責感應血糖、釋放胰島素來平衡血糖水平。但在糖尿病的進展過程中,這些 β 細胞不只是功能變差,甚至會逐漸死亡。這背後有兩個重要的元兇:
當粒線體功能異常或身體處在高血糖、高脂肪的狀態時,會產生大量「活性氧」(ROS),這些自由基就像細胞內的毒氣,會攻擊 β 細胞的結構與 DNA,導致功能受損,甚至細胞死亡。正常情況下,鈣離子是刺激β細胞釋放胰島素的重要訊號。但當細胞內鈣離子過多或控制失調時,就會干擾細胞內部機制,讓細胞無法正常工作,甚至提早進入「細胞凋亡」(細胞自殺)模式。
β 細胞就像精密的工廠,但當自由基太多、鈣離子失控,它們就會「過勞報廢」,讓胰島素分泌能力越來越差。這是糖尿病惡化的一大關鍵。
C. 肥胖與代謝症候群
粒線體功能障礙也與肥胖 [33]和代謝症候群 [34]相關。長期的營養過剩和缺乏運動的生活方式會引發低度慢性發炎(稱為代謝性發炎)和粒線體功能障礙 [33]。脂肪組織中的MD會影響脂肪因子的分泌和發炎狀態,進一步影響全身代謝 [13]。
D. 粒線體功能障礙與胰島素阻抗 - 因果之辯
關於 MD 是 IR 的主要原因,還是繼發於肥胖、高血糖、脂毒性和發炎等因素的結果,這一爭論已持續多年 [29]。
1. 支持 MD 導致 IR 的證據:
· 遺傳學研究顯示某些mtDNA突變可直接導致糖尿病 [35]。
· 研究發現,T2D 患者的體型偏瘦、胰島素抵抗的後代,即使在糖尿病前期,其肌肉中也已存在粒線體功能/生物合成基因表達下降,提示可能存在先天缺陷 [29]。
· 在動物模型中,透過基因操作損害粒線體功能通常會導致胰島素阻抗[29]。
· 機制上,粒線體脂肪酸氧化受損導致的脂毒性物質積累,已被證明會干擾胰島素訊號通路 [29]。
2. 支持 IR /代謝壓力導致 MD 的證據:
· 高血糖、高脂肪酸(脂毒性)和發炎症狀態(常見於肥胖和T2D)本身就已知會損害粒線體,抑制OXPHOS,增加ROS產生,並促進粒線體分裂 [13]。
· 胰島素本身也可能影響粒線體功能,因此IR狀態可能直接對粒線體產生負面影響 [13]。
· 一些研究在某些IR狀態下並未發現明顯的 MD,或者改善胰島素敏感性後 MD 並未完全糾正 [29]。
· 骨骼肌巨大的備用呼吸能力讓人質疑,在許多肥胖和 IR 個體中觀察到的 MD 程度是否足以成為導致 IR 的根本原因 [29]。
3. 綜合觀點 - 可能的雙向強化:
與神經退化性疾病類似,MD 與 IR 之間的關係很可能是複雜且雙向的 [13]。可能存在一個初始的缺陷或脆弱性(遺傳易感性、早期環境暴露)損害了粒線體的基礎功能 [29]。隨後的代謝壓力(如肥胖、營養過剩、缺乏運動)超出了粒線體的代償能力,導致顯著的 MD(脂毒性、ROS 增加),而這種 MD 又反過來積極推動和加劇了 IR [13]。同時,已經建立的 IR 狀態以及伴隨的高血糖、高血脂環境,又會持續對粒線體造成損害 [13]。
這就形成了一個惡性循環,共同促進 T2D 的發生和發展。未來需要更多利用如孟德爾隨機化等方法的研究來進一步釐清因果關係 [36]。
「代謝不靈活性」[30] 是 IR 背景下 MD 的一個關鍵功能性後果。這種由於氧化能力受損而導致的燃料來源(葡萄糖 vs. 脂肪)切換能力下降,直接促成了高血糖(葡萄糖處理能力下降)和脂毒性(脂肪燃燒能力下降)。健康的代謝需要在進食/禁食狀態或運動時切換葡萄糖和脂肪的氧化 [30]。
T2D / IR中的 MD 涉及氧化脂肪和碳水化合物的能力雙雙下降 [13]。這種氧化能力的下降阻礙了有效的燃料切換,導致「代謝不靈活性」[30]。在進食或高血糖狀態下,葡萄糖氧化受損導致高血糖。在禁食或高脂肪狀態下,脂肪氧化受損導致脂質積累(如肌內脂肪、神經醯胺)和脂毒性 [29]。因此,代謝不靈活性是 MD 的直接功能後果,它將粒線體底物處理能力的損害與 IR 和 T2D 的核心病理生理特徵聯繫起來。
粒線體動力學(特別是碎片化)在IR / T2D中也扮演著重要角色 [13],可能作為連接代謝壓力(如高血糖)和下游粒線體功能障礙(如ROS產生、訊號受損)的橋樑。
糖尿病患者肌肉中粒線體更為碎片化 [13],高血糖本身就能誘導碎片化,而碎片化又與ROS增加和胰島素訊號受損相關 [13]。其機制涉及糖尿病環境促進分裂(Drp1激活)並抑制融合(MFN/OPA1水平下降)[13]。這表明粒線體動力學不僅僅是一個被動的標記,而是一個受糖尿病狀態影響並主動加劇 MD 和 IR 的活躍過程。
V. 粒線體功能障礙在心血管疾病(CVDs)中的作用
A. 心臟的能量引擎
我們的心臟是一個從出生到死亡都不停跳動的「永動機」,平均每天跳超過 10 萬次,為全身輸送血液。而要讓這個強力泵浦持續運轉,它需要源源不絕的能量供應。這些能量幾乎全靠粒線體來提供。
心臟的主要燃料是脂肪酸,經由粒線體的「氧化磷酸化」過程轉換成 ATP(能量分子)[2]。在心肌細胞中,粒線體的數量極多,佔了細胞體積的 25 – 30%,遠比其他細胞更多,顯示它們對能量的高度依賴 [17]。
粒線體在心臟裡不只「發電」,還兼職三大任務 [2]:調控鈣離子平衡--鈣離子是心臟跳動節奏的調節器,粒線體幫助穩定它的濃度,避免失控;控制自由基(ROS)--粒線體是 ROS 的主要產生處,也負責清除過量自由基,避免心臟受損;決定細胞存亡 — 當心肌細胞受傷或壓力過大時,粒線體會參與啟動「細胞自殺機制」(凋亡),防止細胞異常增生或癌化。
不只心臟,血管也靠它維持健康 [2],包含血管內皮細胞(控制血管擴張與通透性)及血管平滑肌細胞(負責血管收縮與壓力調控)。這些細胞的正常功能,同樣離不開粒線體的能量與抗氧化支援。一旦粒線體出問題,不只是心臟會「斷電」,連血管也會「失控」,導致高血壓、動脈硬化等問題。
B. 特定疾病關聯
1. 心臟衰竭(Heart Failure, HF):
無論是高血壓、冠心病、心肌病變還是其他病因造成的心衰竭(Heart Failure, HF),科學家發現一個共同的關鍵問題——粒線體功能障礙(Mitochondrial Dysfunction, MD)[2]。可以想像,心臟就像一座永不關機的發電站,而粒線體就是裡頭的核心發電機。一旦這些發電機出問題,整座城市(也就是身體)就會陸續出現故障。在心衰竭中,粒線體的異常表現包含 [2]:
· 能量虧損(ATP不足)——「能量飢餓」假說
心臟是一個能量需求極高的器官,當粒線體無法產出足夠ATP時,就如同油箱見底,心肌無力收縮與舒張。
· 自由基(ROS)過多
受損的粒線體會釋放大量自由基,造成細胞內部氧化壓力,破壞心肌組織,形成惡性循環。
· 粒線體DNA(mtDNA)損傷
粒線體內的基因也可能被破壞,使得它們更難自我修復與維持穩定運作。
· 粒線體動力學失衡(過度分裂)
健康的粒線體會持續進行融合與分裂,但心衰竭患者的粒線體往往過度分裂、難以融合,導致功能碎片化。
· 鈣離子調控異常/過載
心肌的收縮需要精密的鈣離子調控,但粒線體功能失衡會讓鈣離子「淹沒」細胞,引發不穩定跳動與細胞壞死。
· 粒線體生物合成減弱
新粒線體的生成能力下降,導致細胞無法補足損壞的「舊發電機」。
· 細胞凋亡增加 / mPTP 開放
當壓力過高時,粒線體內的通道(如mPTP)會異常打開,啟動細胞死亡程序,使心肌損耗加速。
更值得注意的是,這些粒線體的問題不僅發生在心肌細胞,還出現在骨骼肌中。這也是為什麼心衰竭患者即使靜止時沒症狀,一旦走路、爬樓梯、稍微活動就容易感到疲勞、喘不過氣——因為連肌肉也失去了「能量供應」[37]。心衰竭的根源並不只在於心臟「跳不動」,而是整個粒線體系統陷入崩壞。從能量生產、自由基平衡,到細胞命運決定,粒線體都是背後的操盤者。
能量供應不足直接損害心臟的收縮和舒張功能 [2]。過量的 ROS 會損傷細胞結構,促進心肌重塑(remodeling)[2]。鈣離子超載易引發心律不整並觸發細胞死亡 [2]。粒
線體過度分裂也與細胞凋亡增加有關 [2]。粒線體釋放的 DAMPs(如 mtDNA)或過量 ROS 可引發或加劇心肌炎症 [2]。雖然衰竭的心臟會發生代謝重編程(從依賴脂肪酸轉向更多利用葡萄糖),但糖解產生的 ATP 遠不足以彌補 OXPHOS 的不足 [38]。氧化還原失衡、鈣離子紊亂、炎症反應等形成惡性循環,共同推動 HF 的進展 [3]。
2. 缺血性心臟病/缺血再灌注損傷(Ischemia-Reperfusion Injury, IRI)[2]:
心肌梗塞時是因為血流突然中斷,導致心臟細胞缺氧。但你知道嗎?
當血流重新恢復時(稱為再灌注),反而可能對心臟造成「第二次傷害」,這就叫做「再灌注損傷(IRI)」。在缺血期間,心肌細胞的能量工廠 —— 粒線體失去氧氣,無法產生ATP,開始累積有害物質如自由基(ROS)。這時細胞就像「快撐不住」的引擎。但更大的衝擊,來自於再灌注那一刻:
▼ 氧氣大量湧入 → 粒線體產生爆量ROS(「自由基風暴」)
▼ 鈣離子大量進入細胞 → 粒線體內鈣離子超載
▼ 酸鹼值(pH)劇變→ 粒線體內部環境驟變
這些刺激會打開一個「死亡開關」——粒線體通透性轉換孔(mPTP)。
mPTP打開後會發生粒線體腫脹破裂、細胞能量工廠癱瘓、促凋亡因子釋放及心肌細胞開始壞死或凋亡。也就是說,粒線體的失控,是再灌注損傷的中心機制,甚至導致的細胞死亡比缺血本身還嚴重!研究者正積極尋找保護粒線體的方法,來減輕再灌注傷害,例如:
· 抑制 mPTP 開放的藥物(如Cyclosporin A)
· 強化抗氧化系統
· 調節鈣離子流動
· 使用粒線體靶向抗氧化劑
3. 高血壓(Hypertension)[2]:
高血壓不只是「鹽吃太多」或「壓力太大」造成的。近年研究發現,粒線體功 MD 也是高血壓的幕後推手之一,透過破壞血管健康,悄悄讓血壓節節上升。在我們的血管裡,有兩種關鍵細胞負責維持正常血壓:
▼ 內皮細胞:血管內層的「潤滑膜」,負責釋放一氧化氮(NO),讓血管放鬆、血壓下降。
▼ 血管平滑肌細胞(VSMCs):控制血管的「鬆緊肌」,收縮會讓血管變窄、壓力上升。
當粒線體功能異常、特別是產生過多的活性氧(ROS)時,這兩種細胞就會「失控」:在內皮細胞中,ROS 會清除 NO,讓這個「放鬆訊號」變少,導致血管無法正常擴張,結果就是內皮功能失常、血管緊繃、血壓升高。在血管平滑肌細胞中,ROS會引發慢性發炎,讓細胞變得更大(肥大);促進細胞外基質沉積,讓血管壁變厚、變硬,導致血管結構重塑,失去彈性,讓血壓更難調節。
粒線體功能失調就像是血管裡的「系統性壓力製造者」:不只讓血管變緊、變硬,還讓本來應該放鬆的訊號(NO)失靈。所以,守護粒線體健康,也是在守護血壓的穩定與血管的彈性。
4 .動脈粥樣硬化(Atherosclerosis):
我們都知道,動脈粥樣硬化是造成心肌梗塞、中風等重大心血管疾病的主因。它的特徵是血管內形成脂肪斑塊,讓血流變窄、血壓升高、甚至完全阻塞。近年研究發現:這些斑塊的形成過程,MD 也扮演了重要角色 [8]。當粒線體受損時,會釋放兩種「促炎物質」:
▼ 過多的自由基(ROS):這些氧化分子會攻擊血管細胞,引發局部發炎;
▼ 線體 DNA(mtDNA)片段作為DAMPs 粒:這些原本應該藏在粒線體內的DNA,一旦釋放出來,就會被免疫系統當成「危險訊號」,進一步刺激發炎反應。
這些發炎反應會讓血管內皮變得不穩定、吸引更多免疫細胞與膽固醇堆積,加速形成動脈壁上的粥樣斑塊,也讓斑塊變得更大、更脆弱、更容易破裂,大大提高心血管事件風險 [2]。粒線體不只關係到細胞能量,它的失衡也可能成為促進血管發炎、動脈硬化的關鍵角色。
VI. 粒線體功能障礙在癌症中的作用
A. 粒線體與惡性腫瘤:超越瓦氏效應
提到癌細胞,大多數人第一個聯想到的是它們瘋狂生長、不受控制地分裂。而要支持這種不斷增生,癌細胞的能量來源與正常細胞其實大不相同 —— 這背後的主角之一,就是我們細胞中的能量工廠:粒線體(mitochondria)。早在100年前,科學家瓦爾堡(Warburg)就發現:癌細胞即使有氧氣,也偏好使用「糖解作用」(把葡萄糖變成乳酸)這種效率較低的方式來產生能量。這個現象被稱為瓦氏效應(Warburg effect),當時大家以為這是因為癌細胞的粒線體壞掉了 [14]。現代科學打破了這個迷思。研究發現 [14]:多數癌細胞的粒線體其實沒有壞,只是它們被「重新編程」來配合癌細胞的生長需求。粒線體在癌細胞中依然參與能量產生、合成物質、抗氧化防禦和訊號調控,是癌細胞存活不可或缺的幫兇。
在癌症發展過程中,粒線體可能發生以下變化 [12]:
▼ 基因突變:包括粒線體 DNA 或核 DNA 的變異,會改變粒線體運作方式
▼ 代謝路徑改造:粒線體可能偏好某些能量來源(如脂肪酸或胺基酸)來支持快速生長
▼ 訊號傳導失控:例如幫助癌細胞抵抗凋亡(不正常細胞的自我毀滅機制)
▼ 氧化還原平衡改變:維持癌細胞在高壓環境下的生存能力
雖然癌細胞的代謝方式和正常細胞不同,但它們依然依賴粒線體來完成生長、繁殖和逃避免疫系統的追擊。這也意味著,粒線體可能是治療癌症的一個重要靶點 —— 科學家們正努力尋找「如何斷電癌細胞的能量工廠」。
B. 功能障礙與癌症的聯繫機制
1. 代謝重編程(Metabolic Reprogramming)[12]
儘管癌細胞的糖解作用通常上調(瓦氏效應),但粒線體代謝(特別是三羧酸循環,TCA cycle)對於提供生物合成前體(如檸檬酸用於脂肪合成、TCA 中間產物用於非必需胺基酸合成)、維持氧化還原平衡(如產生 NADPH)以及產生 ATP 仍然至關重要。特定的粒線體功能障礙(如TCA循環酶缺陷)會迫使癌細胞進行代謝適應,例如依賴穀氨醯胺進行還原性羧化來補充 TCA 循環。
2. TCA 循環酶缺陷
IDH1/2(異檸檬酸脫氫酶)、SDH(琥珀酸脫氫酶)和FH(延胡索酸水合酶)基因的突變已被證實是某些癌症(如膠質瘤、副神經節瘤、腎癌等)的致癌驅動因素 [12]。這些突變導致「致癌代謝物」(oncometabolites)如D-2-羥基戊二酸(D-2HG)、琥珀酸和延胡索酸的異常積累。這些代謝物會抑制依賴α-酮戊二酸的雙加氧酶(如TET家族DNA去甲基化酶、組蛋白去甲基化酶KDM)和脯氨醯羥化酶(PHDs),從而引發廣泛的表觀遺傳失調(如DNA和組蛋白高甲基化)和偽缺氧狀態(HIF1α 穩定化),最終促進腫瘤的發生和發展 [12]。
3. mtDNA 突變
體細胞 mtDNA 突變在多種人類癌症中非常普遍。這些突變可能影響ETC複合物的功能,導致ROS產生增加,改變細胞代謝,並可能賦予癌細胞選擇性優勢或劣勢 [12]。關於 mtDNA 突變在癌症中究竟是「驅動突變」(driver mutation)還是「過客突變」(passenger mutation)的作用,目前仍存在爭議 [39]。一些特定的 mtDNA 突變被發現與癌症預後或轉移潛力相關 [39]。
4. 氧化壓力 [12]
MD常常導致粒線體ROS產生增加。適度增加的ROS水平可以促進細胞增殖和基因組不穩定性,有利於腫瘤發展;但過高的ROS水平則可能對細胞產生毒性。癌細胞通常會上調抗氧化系統(如透過NRF2通路)來適應和利用這種氧化壓力。
5. 動力學、自噬與生物合成的改變
粒線體的融合/分裂、自噬和生物合成過程在癌症中也常發生失調 [14]。動力學的改變會影響細胞代謝、對凋亡的敏感性以及細胞週期進程 [14]。粒線體自噬的作用較為複雜,既可能抑制腫瘤(清除受損粒線體),也可能促進腫瘤(維持代謝功能、抵抗治療) [14]。粒線體生物合成常受到癌基因(如MYC)的刺激而增強 [12]。
6. 癌基因訊號傳導 [12]
關鍵的癌基因(如RAS、MYC、HIF)和抑癌基因(如p53)能夠直接調控粒線體的功能、代謝和動力學。反過來,粒線體功能障礙(如透過ROS或代謝物)也能影響這些訊號通路的活性,形成複雜的調控網絡。
C. 爭議焦點
1. 最初認為瓦氏效應是由於粒線體呼吸缺陷導致的觀點已被廣泛修正 [14]。現在普遍認為癌細胞的粒線體通常是功能性的。然而,瓦氏效應(即有氧糖解)的確切「功能」或「益處」至今仍未完全闡明,儘管進行了大量研究。
可能的解釋包括:提供更快速的ATP生成速率以滿足快速增殖需求、為生物合成提供大量的碳骨架(如核苷酸、脂質、胺基酸)、維持氧化還原平衡(產生NADPH)、透過產生乳酸調節腫瘤微環境(如促進免疫抑制)或直接參與訊號傳遞等 [40]。瓦氏效應很可能是多種因素共同作用的結果,以支持癌細胞的快速生長和存活 [40]。
2. mtDNA 突變在癌症中的高頻率引發了對其功能意義的疑問 [39]。這些突變是賦予癌細胞選擇性優勢的「驅動突變」,還是僅僅是由於選擇壓力放鬆或突變率增加而隨機累積的、對腫瘤發展無影響甚至有害的「過客突變」[39]
有證據表明某些特定突變可能是驅動性的 [12],而另一些則可能是過客,甚至可能對癌細胞產生輕微的負面影響 [41]。mtDNA的異質性水平(heteroplasmy level)在判斷其功能時至關重要 [41]。準確區分驅動突變和過客突變仍然是一個挑戰 [39]。
D. 評估證據:粒線體在腫瘤發生中的角色
現有證據強烈支持粒線體對於癌細胞的存活和增殖至關重要,其作用遠超瓦氏效應的最初假設 [14]。特定TCA循環酶突變作為癌症驅動因素的角色已得到充分確立 [12]。癌基因/抑癌基因對粒線體功能的調控作用也相當明確 [12]。然而,關於常見體細胞mtDNA突變的功能影響,證據尚不一致且存在爭議 [39]。
雖然 Warburg 最初提出 MD 是癌症的「原因」[42],但目前的觀點更為細緻。
代謝酶基因(如IDH, SDH, FH)的突變證明,特定的粒線體通路缺陷確實「能夠」啟動腫瘤發生 [12]。然而,在許多其他癌症中,MD 更可能是在腫瘤發生發展「過程中」由於癌基因訊號、缺氧環境或突變累積而「產生」的,然後反過來「促進」腫瘤的進一步發展、轉移和治療抵抗 [12]。這種關係是複雜且依賴於具體癌症類型和背景的。
關於 mtDNA 突變的「驅動者 vs. 過客」之辯,可能受到異質性水平的影響。低水平的突變可能確實是無功能的過客。然而,某些特定的突變如果能達到較高的異質性水平(可能經過了正向選擇),就可能扮演驅動者或調節者的角色,例如透過輕微改變ROS水平或代謝流來影響癌細胞表型,而不是像導致嚴重 PMD 的高異質性突變那樣引起災難性的功能衰竭 [41]。這種觀點有助於調和 mtDNA 突變的高頻率與癌細胞需要功能性粒線體之間的矛盾。
此外,「過客突變」(包括mtDNA過客突變)可能具有「集體負面效應」的概念 [43],為驅動者/過客之辯增添了新的維度。大量累積的、個體效應微弱甚至輕微有害的過客突變,可能最終會損害癌細胞的整體適應性(fitness),在癌細胞的進化過程中形成與驅動突變之間的「拉鋸戰」,甚至可能提供治療的契機(例如,透過進一步增加突變負荷來引發「突變崩潰」)[43]。這意味著基因組不穩定性對癌症而言是一把雙刃劍:它加速了有利驅動突變的積累,但也帶來了有害的過客突變負擔,後者可能抵消甚至超過驅動突變帶來的益處 [43]。
VII. 粒線體功能障礙在慢性疲勞症候群(ME/CFS & PCS)中的作用
A. 連接疲勞與細胞能量學
疲勞是一種複雜的主觀感受,患者常描述為缺乏能量、精神或身體疲憊、耐力下降以及體力活動後恢復時間延長 [16]。肌痛性腦脊髓炎/慢性疲勞症候群(Myalgic Encephalomyelitis/Chronic Fatigue Syndrome, ME/CFS)和新冠後遺症(Post-COVID Syndrome, PCS)是兩種以嚴重、持續性疲勞為核心症狀的疾病。由於粒線體是細胞主要的能量產生單位,MD被認為是解釋這些疾病中疲勞症狀的一個合理的生物學機制 [16]。
事實上,疲勞也是原發性粒線體疾病的標誌性症狀之一 [44]。
B. 在 ME/CFS 和 PCS 中的證據
多項研究提示 ME/CFS 患者存在粒線體功能受損 [45]。部分研究報告患者外周血單核細胞(PBMCs)或肌肉組織的ATP產生/水平下降,暗示存在代謝缺陷(如OXPHOS受損或上游代謝異常) [45]。其他研究觀察到PBMCs的粒線體呼吸功能改變 [46]。然而,研究結果存在矛盾,例如有研究發現非粒線體來源的ATP產生反而增加 [45]。有證據表明ME/CFS患者體內存在DNA和脂質的氧化損傷增加 [16]。
輔酶Q10(CoQ10)水平降低與ME/CFS患者的疲勞嚴重程度持續相關 [44]。肉鹼(Carnitine)水平也常被報導異常,但具體哪種類型的肉鹼以及變化的方向在不同研究中存在差異 [44]。基因表達譜研究發現ME/CFS患者中與粒線體功能(代謝、能量產生等)相關的基因表達存在差異,但涉及的具體基因在不同研究間不盡相同 [44]。一些研究提示可能存在粒線體結構改變 [45],但也有研究報告形態相對正常[45]。最近一項研究發現,與PCS患者相比,CFS患者的肌下膜粒線體(subsarcolemmal mitochondria)形態更不規則、體積更小 [45]。
VIII. 慢性疾病中粒線體功能障礙的匯聚通路
綜合前述討論,粒線體功能障礙在多種看似不同的慢性疾病中呈現出一些共通的核心機制,這些機制相互關聯,共同構成了疾病發展的病理生理基礎。
1. 氧化壓力:
這幾乎是所有討論到的慢性疾病中MD的一個普遍特徵。氧化壓力既是粒線體功能失調的結果,也是進一步損害細胞和組織、推動病理進展的驅動因素。它在神經退化、代謝紊亂、心血管疾病、癌症乃至慢性疲勞中都扮演著重要角色 [12]。
2. 生物能量衰竭(ATP缺乏):
對於能量需求極高的組織(如大腦和心臟)而言,ATP供應不足是MD的直接且嚴重的後果。同時,ATP水平的變化也直接影響代謝調節(如胰島素分泌)和可能導致疲勞感 [2]。
3. 鈣離子恆定失調:
在神經元和心肌細胞等興奮性組織中尤為關鍵,鈣離子失衡會觸發細胞死亡、功能紊亂(如心律不整) [1]。
4. 粒線體動力學與品質控制(自噬)異常:
這是一個日益受到重視的共同機制。融合/分裂失衡和粒線體自噬缺陷影響粒線體網絡的健康、受損胞器的清除以及細胞內訊號傳遞,在多種慢性疾病中都有體現 [2]。
5. mtDNA 損傷/不穩定性:
與衰老、氧化壓力密切相關,是粒線體功能進行性下降的一個重要因素,在癌症和某些遺傳性疾病中尤為突出 [12]。
6. 與發炎的聯繫:
粒線體功能障礙可以觸發或加劇炎症反應(例如,透過ROS、釋放mtDNA DAMPs、激活炎症小體) [2]。反之,持續的炎症狀態也會損害粒線體功能 [38]。這種相互作用可能在許多慢性炎症相關疾病(如T2D、CVD、某些神經退化性疾病)的病理過程中形成惡性循環。
粒線體功能障礙代表了細胞層級穩態的根本性破壞,影響能量供應、氧化還原平衡、訊號傳遞和品質控制等多個核心環節。這使其成為一個合理的共同通路,能夠解釋為何它會參與如此多樣化的慢性疾病的病理過程 [5]。
儘管存在共享的機制,但 MD 的具體表現和後果取決於不同組織的能量需求、代謝特點和脆弱性 [7]。「因」與「果」的辯論在多種疾病中持續存在,且越來越多的證據指向雙向關係和放大迴路。需要利用更先進的研究方法(如孟德爾隨機化 [21]、縱向多組學研究)來進一步釐清。
遺傳背景(nDNA/mtDNA)、環境因素、生活方式和衰老之間的複雜相互作用,使得精確解析 MD 在慢性疾病中的具體作用變得極具挑戰性 [8]。MD 本身的多面向性也增加了研究的複雜度 [5]。
表2:主要慢性疾病中粒線體功能障礙機制匯總
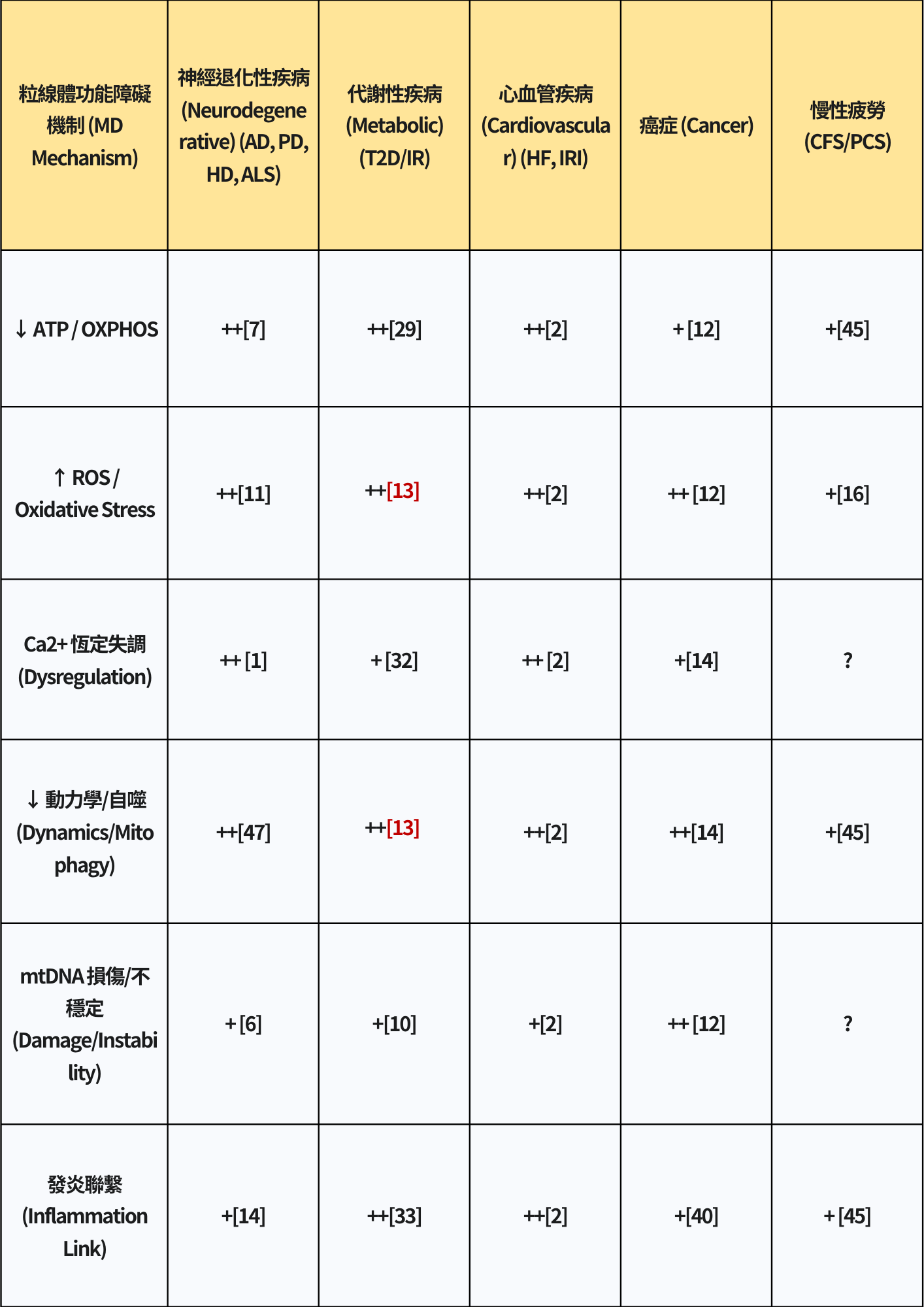
(表格說明:++ 表示證據非常充分/核心機制;+ 表示有證據支持/相關機制;? 表示證據不足或尚不明確)
在許多慢性疾病中,我們常看到一種「雙向惡性循環」的現象。
比方說,在神經退化疾病裡,MD 會加速蛋白質的異常堆積,反過來,這些蛋白聚集又會進一步傷害粒線體;在第二型糖尿病(T2D)中,粒線體問題會加重胰島素阻抗(IR)與脂肪毒性,而這些代謝問題又反過來損害粒線體。
更廣泛地,在許多疾病中,粒線體失衡與慢性發炎之間,也經常互相加劇。
這些現象顯示,粒線體健康與細胞內其他關鍵系統(像是蛋白質維持機制、代謝訊號調控、免疫反應)是緊密交織的。當其中一個環節被破壞,很容易引發其他系統連鎖失衡,最後形成自我延續的惡性循環,加速疾病惡化。
因此,如果要有效阻斷這些惡性循環,單靠針對單一環節的治療可能不足。即便短期改善了一個問題,上游的觸發因素(例如慢性發炎或蛋白錯誤摺疊),或下游造成的傷害,仍可能持續存在。這意味著未來的治療策略,可能需要採取「多靶點」的整合方式,才能真正打破慢性疾病的惡性迴圈。
IX. 治療前景:靶向粒線體以管理慢性疾病
A. 靶向粒線體的理論基礎
鑑於粒線體功能障礙在眾多慢性疾病的病理生理學中扮演著中心角色 [9],將粒線體作為治療靶點提供了一種具有潛在廣泛應用價值的、有前景的治療策略 [2]。這種策略的核心目標是恢復粒線體功能、減少其損傷、改善能量代謝、降低氧化壓力,並最終打破導致疾病進展的病理循環 [2]。
B. 主要治療策略概述
目前正在探索或開發的靶向粒線體的治療策略涵蓋多個方面:
1. 抗氧化劑(Antioxidants):
目標是清除過量的ROS或增強內源性抗氧化防禦能力 [33]。
傳統抗氧化劑如維生素 E、α -硫辛酸(alpha-lipoic acid)[16];輔酶 Q10(CoQ10)[2];N -乙醯半胱胺酸(NAC)[2];以及特異性靶向粒線體的抗氧化劑,如 MitoQ(將 CoQ10 連接到親脂性陽離子 TPP+上以靶向粒線體)[2]、SS-31(一種可穿透粒線體膜的短肽)[35];以及 SOD 或過氧化氫酶(CAT)的模擬物[23]。
儘管在臨床前研究中顯示出希望,但許多抗氧化劑在臨床試驗中的效果往往不佳或結果不一致。這可能歸因於藥物遞送效率低、劑量選擇不當、干預時機太晚,或是因為 ROS 在細胞中同時扮演著損傷者和必需訊號分子的雙重複雜角色 [6]。
2. 代謝調節劑(Metabolic Modulators):
目標是改善粒線體的底物利用效率、提高 ETC 功能或繞過有缺陷的代謝步驟 [2]。
提升細胞內 NAD+ 水平的策略(如補充其前體煙醯胺核糖苷 NR 或煙醯胺單核苷酸 NMN),NAD+ 是許多代謝反應的關鍵輔酶 [6];提供替代能源底物(如生酮飲食產生的酮體)[6];使用藥物改變心臟等組織的底物偏好(如從脂肪酸轉向葡萄糖,或反之,視具體病理而定);激活能量感應器 AMPK(AMP-activated protein kinase)[33]。常用的降糖藥物二甲雙胍(Metformin)也被認為部分透過影響粒線體功能發揮作用 [35]。
部分策略(如 NAD+ 補充、酮體)在動物模型中顯示潛力 [6],一些代謝調節藥物已用於臨床(如二甲雙胍),但針對粒線體的特異性代謝調節仍在發展中。
3.粒線體動力學調節劑(Mitochondrial Dynamics Regulators):
目標是恢復粒線體融合與分裂之間的平衡,減少有害的碎片化或過度融合 [2]。抑制過度分裂的藥物,如 DRP1 抑制劑(例如Mdivi-1)[2];或促進融合的藥物,如 MFN 或 OPA1 的調節劑 [2]。
目前處於臨床前研究階段,在神經退化性疾病和心血管疾病模型中顯示出潛力 [2]。但需要精確調控,因為過度融合也可能有害(如將受損粒線體鎖在網絡中)[6]。
4. 增強粒線體自噬/品質控制(Enhancing Mitophagy/Quality Control):
目標是促進受損粒線體的清除 [14]。
調節 PINK1/Parkin 途徑的藥物;或使用廣譜的自噬增強劑 [14]。目前處於臨床前探索階段,面臨的主要挑戰是如何實現選擇性清除受損粒線體,避免對健康胞器產生影響 [6]。
5. 抑制 mPTP 開放(Inhibiting mPTP Opening)[2]:
目標是阻止粒線體介導的細胞死亡,尤其適用於急性損傷情況,如心肌缺血再灌注損傷。
環孢素A(Cyclosporine A)、TRO40303,進行了一些臨床試驗,但結果不一,其臨床應用價值仍在評估中。
6. 基因治療(Gene Therapy):
目標是糾正導致粒線體功能障礙的潛在遺傳缺陷,主要針對原發性粒線體疾病,但理論上可能擴展應用 [2]。
遞送正確的核基因拷貝;異位表達(allotopic expression,即在核基因組中表達 mtDNA 編碼的基因);以及直接編輯mtDNA 的技術(如 MitoTALENs、粒線體鋅指核酸酶ZFNs、基於CRISPR的工具) [2]。粒線體捐贈是一種用於輔助生殖的技術,旨在防止母系遺傳的 mtDNA 疾病傳遞給下一代 [48]。基因治療面臨諸多挑戰,特別是將治療性基因有效且安全地遞送到粒線體內。
7. 粒線體移植(Mitochondrial Transplantation):
一種新穎的策略,涉及將健康的、分離的粒線體輸注到受損組織或細胞中。在多種疾病的臨床前模型(包括心血管疾病)中顯示出令人鼓舞的結果,如改善心臟功能、減少梗塞面積 [2]。
8. 生活方式干預(Lifestyle Interventions):
運動和飲食對粒線體健康有顯著影響,是基礎且重要的干預手段 [2]。
規律運動(尤其是有氧運動和抗阻運動)已被證明能促進粒線體生物合成,改善其功能 [2]。
飲食調整,如熱量限制、間歇性禁食、地中海飲食 [2]、低升糖指數飲食 [46]、富含抗氧化物和多酚的食物 [33]等,都可能對粒線體健康產生益處。補充特定營養素如肉鹼、CoQ10、NADH [16]等也可能有效。生活方式干預是預防和管理許多慢性疾病的基石,其對粒線體健康的益處得到廣泛認可。
C. 挑戰與未來方向
儘管靶向粒線體的治療策略前景廣闊,但仍面臨諸多挑戰:
▼ 靶向特異性與遞送: 如何將治療藥物精確地遞送到粒線體內部,同時避免影響細胞內其他部位或產生脫靶效應,是一個關鍵的技術難題。
▼ 異質性: 不同疾病、不同組織甚至不同個體之間的粒線體功能障礙可能存在差異,需要更個人化的治療方法。
▼ 因果關係與干預時機: 在許多慢性疾病中,MD與其他病理過程相互交織,釐清確切的因果關係並確定最佳的干預時機至關重要。過早或過晚干預可能效果不佳。
▼ 生物標記的缺乏: 缺乏足夠靈敏和特異的生物標記物來監測粒線體功能變化和治療反應,限制了臨床試驗的設計和評估[4]。
▼ 安全性: 由於粒線體在細胞中的核心作用,任何干預措施都需要仔細評估其潛在的副作用和長期安全性。
未來的研究方向應包括:深入闡明MD在不同慢性疾病中的精確分子機制;開發更可靠的粒線體功能生物標記物;設計更有效的粒線體靶向遞送系統;進行大規模、設計良好的臨床試驗以驗證各種治療策略的有效性和安全性[4]。結合多組學技術和系統生物學方法,有望更全面地理解粒線體在健康與疾病中的作用,並推動精準粒線體醫學的發展。
X. 結論
粒線體,從「罕見疾病」走向「全民健康」的關鍵角色
過去,粒線體功能異常常被認為只是少數遺傳疾病的問題;但現在,科學家發現,它可能是許多常見慢性病背後的共同關鍵。舉例來說,像是阿茲海默症、帕金森氏症這些神經退化性疾病,第二型糖尿病、肥胖這些代謝疾病,甚至心臟病、部分癌症和慢性疲勞症候群,都可能與粒線體的異常有關。這意味著:保護粒線體健康,可能就是預防與改善慢性病的關鍵之一。
粒線體是細胞的「發電廠」,但當它們受損,會連帶引發一連串的問題,例如:
▼ 能量不足(ATP減少):身體疲憊、器官無力
▼ 氧化壓力上升(自由基過多):傷害細胞、加速老化
▼ 鈣離子失衡:影響肌肉收縮與細胞通訊
▼ 粒線體動態失調:粒線體的合併與分裂出現問題
▼ 粒線體自我清理失效(自噬功能下降):舊的或損壞的粒線體無法被清除
▼細胞死亡訊號異常:引發病變或組織退化
這些機制在不同疾病中雖然表現方式不同,但都暗示著:粒線體可能是各種慢性病的「共通弱點」。目前科學界仍在研究一個重要問題:粒線體功能障礙到底是導致疾病的原因,還是疾病惡化的結果?答案可能是兩者兼具。粒線體可能因遺傳、環境或年齡因素而先受損,讓身體對疾病更敏感;但也可能在疾病過程中,被其他壓力因素(如發炎、蛋白堆積等)進一步破壞。
不管哪一種,一旦粒線體進入「惡性循環」,就容易成為加重病情的關鍵推手。
解方:從抗氧化劑到健康生活
既然粒線體這麼重要,是否有方法可以「修補」或「保養」它呢?目前的研究與治療方向包括:
▼ 藥物與營養干預:如抗氧化劑、代謝調節劑、促進粒線體更新的成分
▼ 新興療法:包括基因治療與粒線體移植,雖然潛力大,但仍在研究階段
▼ 生活型態改變:最實用也最有科學支持的方法之一。規律運動、均衡飲食、減少壓力都能有效提升粒線體功能,是每個人都做得到的保健之道。
未來展望:從粒線體看慢性病的全新視角
粒線體不再只是罕見疾病的關鍵字,而是理解、預防與治療慢性病的新切入點。接下來,科學界需要做的包括:
▼ 釐清粒線體異常與疾病之間的因果關係
▼ 發展更準確的診斷工具與生物標記
▼ 找到有效又安全的治療方法
如果能夠針對粒線體這個「細胞健康核心」進行干預,我們或許有機會真正從根本改善許多影響全球數百萬人健康的慢性疾病。
________________________________________________________________

JoiiSports app 虹映科技創辦人 陳立洋執行長
________________________________________________________________
透過 JoiiSports App 企業運動 app 專屬後台 -
JoiiCare 企業 / 組織健康促進服務平台的管理,不僅體現企業對員工健康的關懷,更可增強企業在社會責任中的形象。
另外企業對員工健康和福祉的承諾將成為一項關鍵評價指標,強化企業在 ESG 報告中的表現,可進一步吸引社會大眾、及投資方的關注和支持。
________________________________________________________________
JoiiSports App形象影片
JoiiCare 健康促進服務平台提倡企業線上揪團運動活動,旨在提升員工的參與度和健康福祉。
我們認為健康計畫的成功不僅在於參與,而在於實現實際的健康改善。我們的目標是幫助企業培養更健康的員工,提供有效的策略和工具,激勵員工做出持久的健康改變。
通過專注於真正的健康成果,我們幫助企業實現健康目標,並打造更具生產力、參與度和韌性的團隊,並協助企業客戶在 ESG 報告中的數據生成與撰寫。
歡迎對於職場健康促進服務有需求的人資、福委、廠護、護理師等填寫表單,立即獲得免費諮詢!
聯絡我們
服務專線:0937-122-660 行銷業務處 駱經理
服務信箱:joiicare@joiiup.com
參考文獻
[1] https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2020.600079/full
[2] https://www.mdpi.com/1422-0067/26/5/1917
[3] https://pmc.ncbi.nlm.nih.gov/articles/PMC6118589/
[4] https://www.researchgate.net/publication/367395255_Laboratory_testing_for_mitochondrial_diseases_biomarkers_for_diagnosis_and_follow-up
[5] https://pmc.ncbi.nlm.nih.gov/articles/PMC7255501/
[6] https://www.mdpi.com/2073-4409/14/4/276
[7] https://pmc.ncbi.nlm.nih.gov/articles/PMC10903104/
[8] https://jhrlmc.com/index.php/home/article/download/604/574/2945
[9] https://pubmed.ncbi.nlm.nih.gov/34990812/
[10] https://www.mdpi.com/1873-149X/32/1/9
[11] https://pmc.ncbi.nlm.nih.gov/articles/PMC3618469/
[12] https://www.mdpi.com/1422-0067/21/16/5598
[13] https://diabetesjournals.org/diabetes/article/73/2/151/154089/Mitochondrial-Dynamics-Diabetes-and-Cardiovascular
[14] https://pmc.ncbi.nlm.nih.gov/articles/PMC3844930/
[15] https://my.clevelandclinic.org/health/diseases/15612-mitochondrial-diseases
[16] https://pmc.ncbi.nlm.nih.gov/articles/PMC4566449/
[17] https://www.researchgate.net/publication/390345379_Mitochondrial_dysfunction_in_cardiovascular_disease_investigating_therapeutic_approaches_to_enhance_patient_outcomes
[18] https://pmc.ncbi.nlm.nih.gov/articles/PMC10903091/
[19] https://pmc.ncbi.nlm.nih.gov/articles/PMC9708372/
[20] https://www.neurology.org/doi/10.1212/WNL.0000000000002705
[21] https://pmc.ncbi.nlm.nih.gov/articles/PMC10823732/
[22] https://www.semanticscholar.org/paper/Mitochondria-dysfunction-and-neurodegenerative-or-Morais-Strooper/d0211f1e1bb6e02699f3eb5613fdbd2085091570
[23] https://pubmed.ncbi.nlm.nih.gov/38241161/
[24] https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2021.617588/full
[25] https://pmc.ncbi.nlm.nih.gov/articles/PMC3226310/
[26] https://academic.oup.com/braincomms/article/1/1/fcz009/5544105
[27] https://www.researchgate.net/publication/390520019_Mitochondrial_Dysfunction_Genetic_Predisposition_and_Targeted_Interventions_in_Neurodegenerative_Diseases_and_Cognitive_Decline_A_Meta-Analysis_of_Mechanisms_and_Treatments
[28] https://pubmed.ncbi.nlm.nih.gov/39367555
[29] https://pmc.ncbi.nlm.nih.gov/articles/PMC4261703/
[30] https://pmc.ncbi.nlm.nih.gov/articles/PMC10135185/
[31] https://elifesciences.org/articles/87340/peer-reviews
[32] https://www.mdpi.com/2218-273X/13/1/126
[33] https://pmc.ncbi.nlm.nih.gov/articles/PMC3603328/
[34] https://pubmed.ncbi.nlm.nih.gov/38744846/
[35] https://pmc.ncbi.nlm.nih.gov/articles/PMC5284490/
[36] https://pmc.ncbi.nlm.nih.gov/articles/PMC11392782/
[37] https://pmc.ncbi.nlm.nih.gov/articles/PMC3855291/
[38] https://www.jci.org/articles/view/120849
[39] https://pmc.ncbi.nlm.nih.gov/articles/PMC6358502/
[40] https://www.researchgate.net/publication/289499814_The_Warburg_Effect_How_Does_it_Benefit_Cancer_Cells
[41] https://pmc.ncbi.nlm.nih.gov/articles/PMC9671861/
[42] https://pmc.ncbi.nlm.nih.gov/articles/PMC4946416/
[43] https://aacrjournals.org/cancerres/article/77/18/4763/622823/The-Damaging-Effect-of-Passenger-Mutations-on
[44] https://pmc.ncbi.nlm.nih.gov/articles/PMC4136529/
[45] https://www.mdpi.com/1422-0067/25/3/1675
[46] https://www.ifm.org/articles/chronic-fatigue-and-mitochondrial-health
[47] https://www.mdpi.com/2227-9059/13/2/327
[48] https://pubmed.ncbi.nlm.nih.gov/27775730/










